中文内容

概览
- 实验验证:通过使用 MatterSim-v1 进行高通量筛选,我们此前将四方相磷化钽(TaP)识别为一种潜在的高性能热导体。现在,我们已通过实验合成了它,并测得其热导率(152 W/m/K)接近硅的热导率。
- 更快的模拟:我们已将 MatterSim-v1 模型推理速度提升了 3-5 倍,并将其与 LAMMPS 软件包集成,从而能够在多个 GPU 上进行大规模模拟。
- 新模型发布:我们正在推出 MatterSim-MT,这是一种用于计算机材料表征的多任务基础模型,能够模拟复杂的、多性质现象,超越了仅靠势能面所能捕捉的范围。
材料设计支撑着从纳米电子学到半导体设计和储能等广泛的技术进步。然而,新型材料的开发周期仍然缓慢且成本高昂。通用机器学习原子间势旨在通过为广泛材料提供准确的稳定性和性质预测来加速材料设计过程。这些模型比传统的第一性原理模拟快几个数量级,将以往不切实际的问题转变为可在数小时内完成的常规计算。自我们推出 MatterSim-v1 模型以来,它因能够在真实条件下(包括有限温度和压力)准确模拟材料而在材料科学界受到欢迎。
今天,我们有几项令人振奋的 MatterSim 更新要分享。其中包括对 MatterSim 在热导体方面预测的实验验证、用于加速模拟的性能改进,以及推出一种用于材料表征的新型多任务基础模型。
实验验证

高热导率材料在热管理中发挥着关键作用,可防止过热并提高能源效率。例如,金刚石、铜和硅等成熟的高导热材料被广泛用于各种冷却应用。设计下一代导热材料可能推动计算、功率电子和航空航天技术的发展。然而,要做到这一点,需要准确预测候选材料的热导率数值。
在固体中,热量主要通过两种方式传递:振动的原子(声子)和运动的电子。声子贡献可以使用机器学习原子间势来估算,从而能够筛选数千种候选材料,在进行昂贵的实验验证之前,将搜索空间缩小到最有前景的材料。
“MatterSim 迄今已生成规模最大的计算热导率数据库。这为探索比以往广泛得多的材料空间打开了大门 […]。”——德克萨斯大学达拉斯分校 Bing Lv 教授
我们与德克萨斯大学达拉斯分校(UT Dallas)、伊利诺伊大学厄巴纳-香槟分校以及加州大学戴维斯分校(UC Davis)合作,使用 MatterSim-v1 筛选了超过 240,000 种候选高热导材料。如图 1(左)所示,MatterSim 的预测与第一性原理模拟具有良好一致性。来自 UC Davis 的 Davide Donadio 教授表示:“MatterSim 模型能够兼具准确性和计算效率,预测热导率这样敏感的性质,这让我感到惊讶。这正是实现如此规模筛选的关键,数十万种晶体如果采用传统方法将完全无法企及。”来自 UT Dallas 的 Bing Lv 教授补充道:“MatterSim 迄今已生成规模最大的计算热导率数据库。这为探索更广泛的材料空间打开了大门
“我们第一次能够在大规模范围内检验关于哪些因素控制热导率的传统理解 […]”——伊利诺伊大学厄巴纳-香槟分校 David Cahill 教授
基于这些预测,我们已将四方相磷化钽(TaP)确定为一种潜在的高导热材料。我们在 UT Dallas 实验合成了四方相磷化钽(TaP),并在 University of Illinois Urbana-Champaign 测量了其热导率(我们最佳样品为 152 W/m/K),接近硅的热导率。虽然我们并非首次合成四方相 TaP,但此前该材料尚未被视为导热材料。这些结果展示了 MatterSim 如何助力功能材料的识别:“我们首次能够大规模检验关于控制热导率因素的传统理解,同时推动发现新的功能材料,在热导率与质量密度、元素丰度和环境稳定性等其他重要约束之间取得平衡,”David Cahill 教授表示。
性能提升
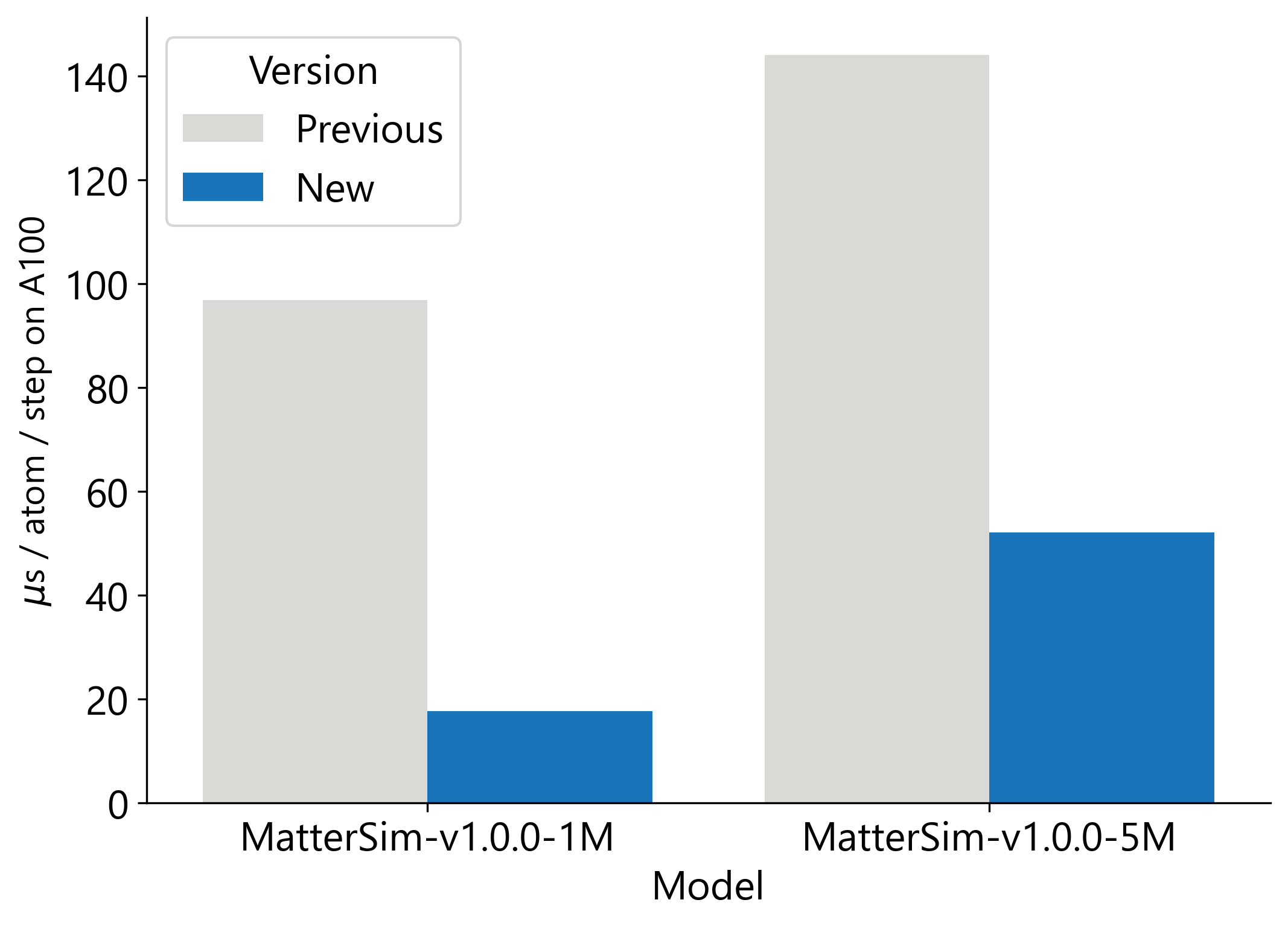
我们通过发布多项开源的性能和可用性改进,使 MatterSim-v1 的速度显著提升。首先,我们结合更快的图构建、提前编译以及减少原子表示之间的转换来加速模型推理,使 MatterSim-v1.0.0-5M 提速 3 倍,MatterSim-v1.0.0-1M 提速 5 倍(见图 2)。为了让 MatterSim-v1 更易于使用,我们已将其集成到广泛使用的 LAMMPS 模拟软件中,使用户能够在现有工作流程中轻松地将模型推理扩展到多个 GPU。
新模型发布
在 MatterSim-v1 成功的基础上,今天我们通过发布 MatterSim-MT 扩展 MatterSim 模型家族:这是一个用于计算机模拟材料仿真和性质表征的多任务(MT)基础模型。该模型能够原生预测能量、力、应力以及若干重要的材料性质。
MatterSim-MT 在超过 3500 万个第一性原理标注的结构上进行了预训练,涵盖 89 种元素、最高 5000 K 的温度以及最高 1000 GPa 的压力。它还在多种性质上进行了进一步微调,包括 Bader 电荷、磁矩、Born 有效电荷和介电矩阵。MatterSim-MT 开箱即用,可作为预测材料结构、动力学和热力学的基础模型。其多任务架构还支持广泛的复杂模拟,而这些模拟无法仅由势能面捕捉。准确模拟这些现象的能力对于催化和储能等应用至关重要。
在此,我们通过三个案例研究展示这些多任务能力:振动光谱、铁电开关和电化学氧化还原。每个示例都需要不同的性质预测组合。在完整手稿中,我们还表明,MatterSim-MT 能够随更多数据和参数实现良好扩展,可以高效微调到更高层次的理论,并可通过主动学习系统性地扩展到新体系。
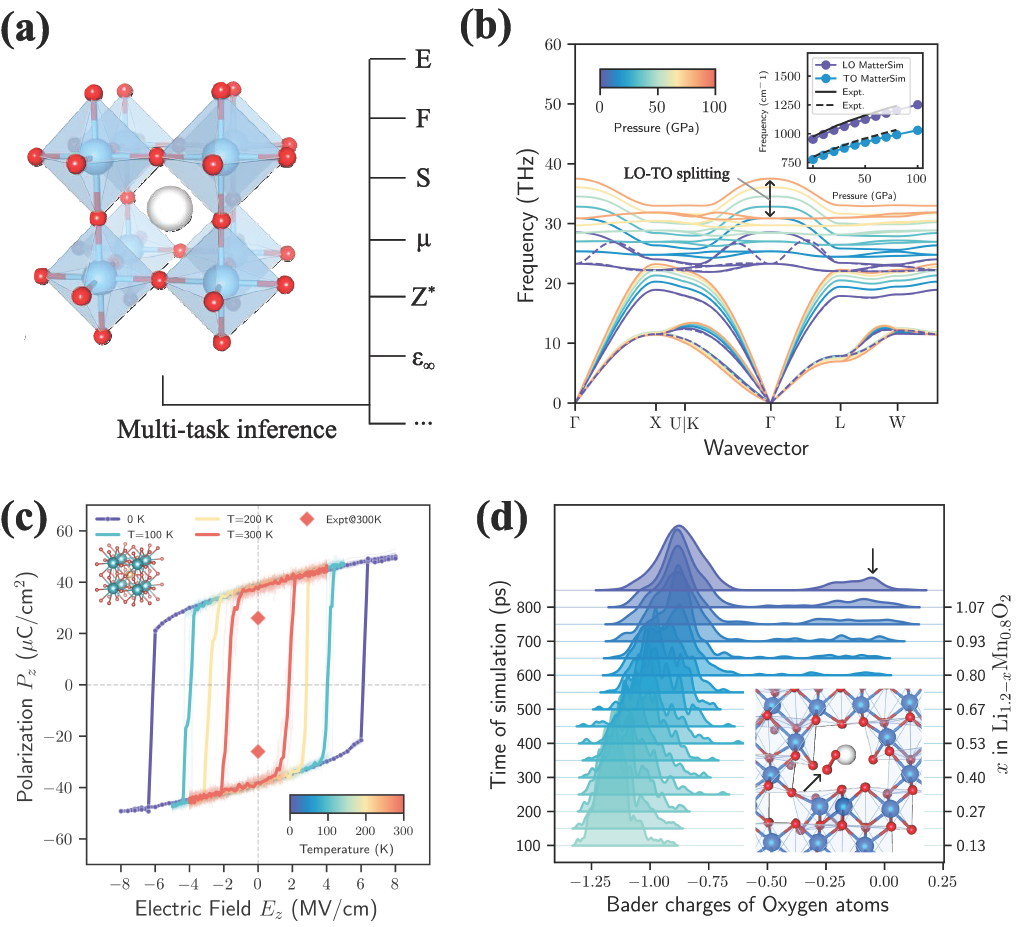
首先,我们关注振动光谱学,这是一种通过测量原子键的自然振动方式来识别物质的技术。我们展示了对 Born 有效电荷和介电性质的预测如何使极性晶体中的声子谱计算成为可能。在这些材料中,带相反电荷的离子彼此相对振动。根据振动方向的不同,这可能导致电荷积累,从而产生宏观电场,将光学声子模式分裂为频率较高的纵向(LO)分支和频率较低的横向(TO)分支。作为案例研究,我们模拟了用于高功率电子器件的材料 3c-碳化硅(3c-SiC)在极端压力下的这种行为。如图 3(b) 所示,MatterSim-MT 预测的 Born 有效电荷与理论值和实验值都非常一致。由此产生的 LO-TO 分裂
预测得到的 Born 有效电荷还使我们能够模拟体系对外加电场的响应。在铁电材料中,离子呈非对称排列,使晶体具有可由外加电场翻转的净电极化。在图 3(c) 中,我们通过模拟外加电场下的钛酸钡(BaTiO3)展示了这一点,再现了其极化的翻转。所得的滞回曲线正确显示,在 300 K 下的有限温度效应会使极化更容易翻转,尽管预测的自发极化(38 μC/cm2)略高于实验值(26 μC/cm2)。这种差异很可能源于底层第一性原理计算中众所周知的欠束缚问题。
最后,我们预测原子电荷,以研究化学键合和氧化还原过程中的电子自由度。我们考察了正极材料 Li1.2 – xMn0.8O2 在模拟电池充电过程中的行为。这些富锂过渡金属氧化物因其高能量密度而成为有前景的下一代电池材料,但存在与阴离子氧氧化还原机制相关的不可逆容量损失问题。我们通过在 1000 K 下进行分子动力学模拟,并逐步提取锂以模拟电池充电,复现了这一现象。我们观察到随时间推移出现明显转变:起初,锰(Mn)原子提供充电所需的电子,但随着更多锂被移除,氧原子被迫转而失去电子(从阳离子氧化还原转变为阴离子氧化还原),这一点表现为 Bader 电荷随时间推移向负性较低的方向移动(图 3(d))。这种失稳
下一步
通过实验验证、显著的性能提升以及新的多任务能力,MatterSim 正朝着在材料设计中更实用、更具决策相关性的应用迈进。这些进展共同帮助材料科学家更快地从大规模计算筛选转向有针对性的实验跟进和与决策相关的科学工作流。我们很期待看到材料科学界如何在各自领域应用这些进展。
随着 MatterSim 在真实世界的材料发现流程中被测试、扩展和集成,我们期待继续开展合作。
致谢
这项工作是由 Microsoft Research AI for Science 牵头,与 Microsoft Research Accelerator 以及 University of Texas Dallas(由 MSR Accelerator 支持)、University of Illinois Urbana-Champaign 和 University of California Davis 的合作者共同开展的高度协作、跨学科努力的成果。本工作的贡献者包括 Han Yang、Xixian Liu、Chenxi Hu、Yichi Zhou、Yu Shi、Chang Liu、Junfu Tan、Jielan Li、Guanzhi Li、Qian Wang、Yu Zhu、Zekun Chen、Shuizhou Chen、Fabian Thiemann、Claudio Zeni、Matthew Horton、Robert Pinsler、Andrew Fowler、Daniel Zügner、Tian Xie、Lixin Sun、Yicheng Chen、Lingyu Kong、Yeqi Bai、Deniz Gunceler、Frank Noé、Hongxia Hao、Ziheng Lu、Zixin Zhai、Mengfan Wu、Haoke Qiu、Mingfa Tang、Tie-Yan Liu、Haiguang Liu、Tao Qin、David G. Cahill、Bing Lv、Davide Donadio、Shoko Ueda 和 Kenji Takeda。
在新标签页中打开








